


USFDA Certification: Everything You Need to Know Before Entering the US Market

The United States Food and Drug Administration is one of the most respected and rigorous regulatory bodies in the world. For any manufacturer, exporter, or business that wants to introduce products — whether food, drugs, cosmetics, medical devices, or dietary supplements — into the American market, obtaining USFDA certification is not just a formality. It is a fundamental requirement that signals safety, quality, and credibility to consumers, healthcare professionals, distributors, and retail giants alike. Understanding what this certification means, how it works, and what it demands of businesses is essential for anyone serious about global trade with the United States.
What Is USFDA Certification and Why Does It Matter
USFDA certification refers to the formal process by which the US Food and Drug Administration evaluates and approves products or facilities to confirm that they meet the federal regulatory standards set by the United States government. The FDA operates under a vast legislative framework, including the Federal Food, Drug, and Cosmetic Act (FD&C Act), the Public Health Service Act, and numerous amendments such as the Food Safety Modernization Act (FSMA) and the Drug Quality and Security Act. These laws collectively empower the FDA to regulate the manufacture, labeling, safety, efficacy, and distribution of a wide variety of products consumed or used by the American public.
The importance of USFDA certification cannot be overstated. In practical terms, products without proper FDA approval or registration face immediate rejection at US ports of entry, legal action, and complete market bans. For pharmaceutical companies, a drug cannot be commercially distributed in the US without FDA approval. For food manufacturers, facilities must be registered with the FDA and comply with FSMA guidelines or face import alerts. For medical device makers, marketing clearance through the 510(k) pathway or premarket approval (PMA) is legally required before a product can be sold. Beyond legal compliance, the certification carries enormous reputational weight — American consumers and healthcare buyers inherently trust FDA-approved or FDA-cleared products more than uncertified alternatives.
The Different Types of USFDA Certification Across Product Categories
USFDA Certification for Food Export from India The FDA does not operate with a single, one-size-fits-all certification system. Instead, the regulatory requirements vary significantly depending on the product category, the risk level it poses to public health, and the nature of its use. Understanding the specific pathway relevant to your product is the first and most critical step in the USFDA certification journey.
Pharmaceutical and Drug Approval Pathways
For pharmaceutical companies, USFDA certification typically involves either a New Drug Application (NDA) or an Abbreviated New Drug Application (ANDA). The NDA pathway applies to brand-new drugs entering the market for the first time, requiring the manufacturer to submit extensive clinical trial data, pharmacological studies, safety information, and proposed labeling. The FDA rigorously reviews this data — a process that can take several years — before granting approval. The ANDA pathway, used for generic drugs, requires manufacturers to demonstrate bioequivalence to an already-approved reference drug, making it a somewhat faster route to market.
Biologics, which include vaccines, blood products, and gene therapies, require a Biologics License Application (BLA), which is equally demanding and involves detailed manufacturing process validation. Over-the-counter (OTC) drugs follow their own monograph system, allowing products that meet established FDA standards to be marketed without individual approval — though they must still comply with labeling and formulation requirements. In all cases, USFDA certification for pharmaceuticals demands that the manufacturing facility itself be inspected and approved, meaning good manufacturing practices (GMP) compliance is inseparable from the product approval process.
Medical Device Clearance and Approval
Medical devices are regulated under a three-class risk system. Class I devices, which pose minimal patient risk, are generally subject to general controls and most are exempt from premarket notification. Class II devices, covering moderate-risk items like infusion pumps and surgical gloves, typically require a 510(k) premarket notification — a process where the manufacturer demonstrates that the new device is "substantially equivalent" to a legally marketed predicate device. Class III devices, which include life-sustaining or life-supporting equipment like pacemakers and cochlear implants, require full Premarket Approval (PMA), the most demanding USFDA certification pathway, requiring valid scientific evidence of safety and effectiveness.
The FDA also offers the De Novo pathway, which serves as an alternative classification request for novel, low-to-moderate risk devices that have no predicate. Understanding which pathway applies, assembling the right technical documentation, and conducting the required clinical evaluations are tasks that require dedicated regulatory expertise. Foreign manufacturers seeking US market access must also appoint a US Agent and register their facility annually with the FDA — failure to do so results in automatic import alerts that block product shipments.
Food Facility Registration and FSMA Compliance
Food manufacturers, processors, packers, and storage facilities that produce or handle food for US consumption must register with the FDA under the Bioterrorism Act of 2002. This registration must be renewed every two years. However, registration is only the first layer of USFDA certification compliance for food businesses. The Food Safety Modernization Act, signed into law in 2011 and fully phased in over subsequent years, transformed food regulation from a reactive model to a preventive one. Under FSMA, food facilities are now required to implement a written Food Safety Plan that includes hazard analysis, preventive controls, a supply-chain program, a recall plan, and corrective action procedures.
Foreign Is USFDA Certification Mandatory for Exporting Food suppliers exporting food to the US must comply with the Foreign Supplier Verification Program (FSVP), which places accountability on US importers to verify that their overseas suppliers meet American safety standards. This effectively exports USFDA certification requirements to production facilities around the world, making it a global compliance standard. Failure to comply with FSVP can result in import refusals, warning letters, and legal liability for both the importer and the foreign supplier.
How to Prepare for USFDA Certification: Key Steps for Manufacturers
Achieving USFDA certification — regardless of the product category — is not something that happens overnight. It requires structured preparation, thorough documentation, and a culture of quality that permeates the entire organization. Businesses that approach the FDA process reactively, rather than proactively, almost always face setbacks that delay market entry and damage business relationships.
Building a Robust Quality Management System
The foundation of any successful USFDA certification effort is a well-implemented Quality Management System (QMS). For pharmaceutical and device manufacturers, this means full compliance with the FDA's Current Good Manufacturing Practices (cGMP) or Quality System Regulation (QSR) for devices. The QMS must cover every aspect of production — from the qualification of raw material suppliers to the final release of finished goods. Standard operating procedures (SOPs), batch records, deviation logs, change control procedures, and comprehensive training programs are not optional extras. They are the evidence that an FDA investigator will review during an inspection to determine whether the facility meets certification standards.
Companies that have obtained ISO 9001, ISO 13485, or ISO 22000 certification often find the transition to FDA compliance smoother because these systems share many structural similarities with FDA requirements. However, ISO certification does not replace USFDA certification — the two systems complement each other, and the FDA has its own specific requirements that go beyond the ISO framework in several areas.
Facility Inspection and Pre-Approval Readiness
Before granting USFDA certification for manufacturing facilities, the FDA typically conducts facility inspections. For pharmaceutical plants, this is called a Pre-Approval Inspection (PAI) and is triggered by Documents Required for USFDA Certification the submission of an NDA or ANDA. Investigators evaluate whether the facility's manufacturing processes, equipment, and documentation align with what was described in the regulatory submission. For food facilities, the FDA conducts routine surveillance inspections and for-cause inspections when problems are reported.
Preparing for an FDA inspection requires systematic gap analysis — identifying where current practices fall short of FDA requirements and implementing corrective measures well in advance. Many companies engage regulatory consultants or hire in-house regulatory affairs specialists to manage this process. Mock inspections, internal audits, and supplier qualification programs are all standard preparation tools. Companies that treat inspection readiness as an ongoing operational discipline rather than a one-time event consistently achieve better outcomes during actual FDA visits.
Common Challenges in Obtaining USFDA Certification
Despite the best intentions and preparations, many manufacturers encounter significant hurdles on the road to USFDA certification. Understanding these challenges in advance allows businesses to plan more realistically and avoid costly mistakes.
One of the most frequent difficulties is inadequate documentation. The FDA operates on the principle of "if it isn't written down, it didn't happen." Vague SOPs, incomplete batch records, missing validation data, or poorly structured technical files can result in a Complete Response Letter (CRL) for drug applications, a Request for Additional Information (RAI) for device submissions, or an Untitled or Warning Letter for food facility violations. Each of these responses prolongs the certification timeline and adds cost to the process.
Another common challenge is managing the intersection of US regulations with those of the manufacturer's home country. Companies operating under the European CE marking system, India's CDSCO regulations, or China's NMPA requirements often find that the FDA requires additional or different types of evidence than they are accustomed to providing. The USFDA certification process requires applicants to adapt their documentation to a specifically American regulatory language and framework, which can be unfamiliar and demanding for companies new to the US market.
Supply chain complexity also presents challenges, particularly under FSMA's FSVP rule and the drug supply chain security requirements of the Drug Supply Chain Security Act (DSCSA). Traceability, supplier verification, and chain-of-custody documentation are increasingly scrutinized by the FDA, especially in the wake of high-profile contamination events and counterfeit drug scandals that have underscored the importance of end-to-end supply chain integrity.
The Role of Regulatory Consultants in USFDA Certification
Given the complexity and high stakes of USFDA certification, many organizations — especially small and medium-sized enterprises entering the US market for the first time — choose to work with experienced regulatory affairs consultants or contract research organizations (CROs). These professionals bring deep familiarity with FDA requirements, established relationships within the regulatory community, and practical experience navigating the submission and inspection process.
A good regulatory consultant will help a company choose the correct approval pathway, prepare and review submissions for completeness and accuracy, coordinate with the FDA during the review process, manage responses to deficiencies or additional information requests, and prepare the facility for inspection. While this expertise comes at a cost, it is almost always a worthwhile investment when measured against the expense and delay of a failed submission or a rejected import shipment.
Maintaining Compliance After USFDA Certification
Obtaining USFDA certification is a significant achievement, but it is not the end of the regulatory journey — it is the beginning of an ongoing compliance obligation. The FDA requires post-market surveillance for drugs and devices, periodic safety update reports, adverse event reporting, and continuous adherence to GMP or QSR standards. For food businesses, FSMA compliance is a living process that demands regular review and updating of the Food Safety Plan, ongoing monitoring of preventive controls, and documented corrective actions whenever deviations occur.
The FDA also conducts periodic re-inspections of approved manufacturing facilities, and any major changes to products, processes, equipment, or facilities may require prior approval or notification to the agency. Companies that maintain robust internal compliance programs and treat regulatory requirements as integral to their business operations rather than as an external burden consistently avoid the costly and reputationally damaging experience of FDA warning letters, consent decrees, or product recalls.
Frequently Asked Questions About USFDA Certification
What is the difference between FDA registration and FDA approval?
FDA registration refers to the process by which a facility or establishment formally notifies the FDA of its existence and the products it manufactures or processes. It is primarily an administrative requirement. FDA approval, on the other hand, is a substantive review process in which the FDA evaluates the safety, efficacy, and quality of a specific product before it can be legally marketed in the United States. Both are components of USFDA certification, but they serve different purposes and apply at different levels — the facility versus the product.
How long does the USFDA certification process take?
The timeline varies widely depending on the product category and the regulatory pathway. A standard 510(k) medical device submission typically takes three to twelve months for FDA review. An NDA for a new pharmaceutical can take ten to twelve months under the standard review track, or six months under the priority review track for products addressing serious conditions. Generic drug ANDAs have historically taken several years, though recent FDA initiatives have worked to reduce this backlog. Food facility registration can be completed in a matter of days, but full FSMA compliance readiness may take months to implement for companies starting from scratch.
Is USFDA certification mandatory for exporting to the United States?
For most regulated product categories — pharmaceuticals, medical devices, and food — yes, some form of FDA regulatory compliance is legally required before products can be imported into or sold in the United States. The specific requirements depend on the product type. Unapproved drugs cannot be commercially marketed, and they face automatic detention at US ports. Food imports are subject to FSMA rules and FSVP obligations. Medical devices without proper premarket clearance or approval are considered adulterated or misbranded under US law and cannot be legally sold.
Can small businesses afford USFDA certification?
The FDA offers several programs to assist small businesses, including reduced user fees for small drug manufacturers, the CDER Small Business and Industry Assistance (SBIA) program, and pre-submission meetings that allow device manufacturers to clarify expectations before investing in a full submission. While USFDA certification is undeniably resource-intensive, the investment typically pays off in access to the world's largest and most lucrative healthcare and consumer market.
What happens if a company fails an FDA inspection?
If the FDA identifies significant violations during an inspection, it issues a Form 483 — a list of inspectional observations — at the close of the inspection. Companies are expected to respond promptly with a corrective action plan. In serious cases, the FDA may issue a Warning Letter, which is publicly posted and requires a formal response. In extreme cases involving ongoing significant violations, the FDA can pursue consent decrees, seizures, injunctions, or criminal prosecution. For facilities seeking USFDA certification for the first time, a failed inspection delays approval and may require a re-inspection before the process can proceed.
Conclusion
The path to USFDA certification is demanding, detailed, and deeply consequential for any business that aspires to participate in the US market. It demands not just paperwork and submissions, but a genuine organizational commitment to product quality, consumer safety, and regulatory transparency. Whether a company is a pharmaceutical giant filing an NDA for a breakthrough therapy, a mid-sized food processor seeking FSMA compliance, or a medical device startup navigating the 510(k) process for the first time, the journey toward USFDA certification shapes how a business thinks about its products and its responsibilities to the people who use them.
The FDA's rigorous standards exist for good reason — they protect hundreds of millions of American consumers from unsafe products every year. Businesses that embrace these standards rather than merely tolerate them build stronger products, more resilient operations, and more trusted brands. In the end, USFDA certification is not simply a regulatory checkpoint. It is a declaration of quality and a commitment to excellence that resonates across the global marketplace.
Advertisment
Categories
Read More
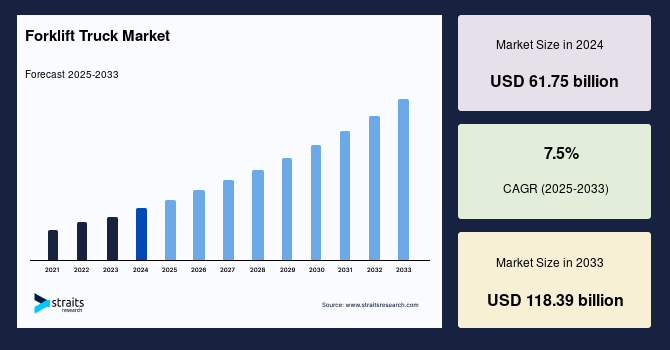
Forklift Truck Industry Insights: Straits Research recently introduced the latest update on the Forklift Truck Market that provides an extensive outlook of the market, analyzing key growth opportunities, challenges, risk factors, and emerging trends across diverse geographic regions. The report offers a definitive and meticulous analysis of the Forklift Truck industry size,...
🔴📺📱👉 CONTINUE WATCHING... https://ns1.iyxwfree24.my.id/movie/EW5 Trending in the US: The Rise of Leaked Content and What It Means for Fans As the world of adult content continues to evolve, fans are increasingly turning to leaked material for a glimpse into the lives of their favorite creators. One recent development has sparked intense curiosity: the emergence of leaked content from...
🔴📺📱👉 CONTINUE WATCHING... https://ns1.iyxwfree24.my.id/movie/bRkh [-wATCH-]—The Viral Sensation Sweeping the Nation: What's Behind the Buzz? In today's digital landscape, it's not uncommon for certain videos to catapult to fame overnight, captivating audiences and sparking conversations on social media. One such phenomenon has been taking the US by storm, with users scrambling to...
✅ CLICK HERE TO STREAMING https://ns1.iyxwfree24.my.id/movie/b3lF BREAKING: Ice Spice's Private Messages Leaked on Discord, Leaving Fans Reeling The Lead In a shocking turn of events, a series of private messages allegedly belonging to rapper Ice Spice have been leaked on the online platform Discord. The leaked messages, which have been circulating on social media, have left fans and...
🌐 CLICK HERE 🟢==►► WATCH NOW 🔴 CLICK HERE 🌐==►► DOWNLOAD NOW https://ns1.iyxwfree24.my.id/movie/b2Jn BREAKING: Lanhxinhyeu Clip 06 No Sensor Sparks Social Media Frenzy The Lead: What's Behind the Buzz? A clip from lanhxinhyeu's latest content, specifically clip 06, has taken the internet by storm, with fans and non-fans alike going wild on social media. The clip, which has been...